This section evaluates whether species’ environmental preferences inferred from the JSDM are phylogenetically structured. We first quantify global phylogenetic signal in each species-level coefficient using Pagel’s λ, then characterise how this signal varies across phylogenetic distances using phylogenetic correlograms. Finally, we use Local Indicators of Phylogenetic Association (LIPA) to identify clades in which closely related species show unusually similar (or unusually dissimilar) preferences.
To relate model outputs to evolutionary history, we combine two data sources: (i) posterior summaries of species-specific environmental coefficients (regional climate, geomorphological subtypes, and SWI), and (ii) a backbone seed-plant phylogeny. The workflow below harmonises taxonomy between the French Guiana checklist and the megatree, constructs a phylogeny for the analysed species, and matches each tip to the corresponding coefficient values.
Code
Species_list_path <-here_rel("data", "inventory_data", "Species_list.csv")Summary_beta <- vroom::vroom(here_rel("results", "models", "H1", "beta_fitH1.tsv.gz")) |>filter(Scale %in%c("Regional", "Landscape", "Local")) |>mutate(# significance at 80% CIsig80 = (l >0& h >0) | (l <0& h <0),sign =case_when( sig80 & m >0~"positive", sig80 & m <0~"negative",TRUE~"ns" ) ) |>mutate(species =paste(genus, species)) |>select(species, m, parameter) |>spread(parameter, m)## Rows: 9104 Columns: 10## ── Column specification ────────────────────────────────────────────────────────## Delimiter: "\t"## chr (5): variable, Scale, parameter, genus, species## dbl (5): m, ll, l, h, hh## ## ℹ Use `spec()` to retrieve the full column specification for this data.## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.Species_list <-read_csv2(Species_list_path) |>rename(SpEpithet = specie, species = taxonomic_name) |>select(species, genus, family, selected) |>filter(species %in% Summary_beta$species, !duplicated(species))## ℹ Using "','" as decimal and "'.'" as grouping mark. Use `read_delim()` for more control.## Rows: 1726 Columns: 7── Column specification ────────────────────────────────────────────────────────## Delimiter: ";"## chr (5): taxonomic_name, family, genus, specie, ID.TAXREF## dbl (2): ID, selected## ℹ Use `spec()` to retrieve the full column specification for this data.## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.if (file.exists(here_rel("data", "inventory_data", "tree_beta.rds"))) { tree_beta <-readRDS(here_rel("data", "inventory_data", "tree_beta.rds"))} else { megatree <-read.tree("https://github.com/megatrees/plant_20221117/raw/refs/heads/main/plant_megatree.tre") gen.list <-read.csv("https://github.com/megatrees/plant_20221117/raw/refs/heads/main/plant_genus_list.csv", sep =",")# Correction for modified genus between megatree / species list (Molino et al. 2022). splist_cor <- Species_list splist_cor$genus <-gsub("Marlimorimia", "Inga", splist_cor$genus) splist_cor$species <-gsub("Marlimorimia", "Inga", splist_cor$species) splist_cor$genus <-gsub("Robrichia", "Enterolobium", splist_cor$genus) splist_cor$species <-gsub("Robrichia", "Enterolobium", splist_cor$species) splist_cor$genus <-gsub("Gwilymia", "Entada", splist_cor$genus) splist_cor$species <-gsub("Gwilymia", "Entada", splist_cor$species) tree_beta <-phylo.maker(splist_cor, megatree, gen.list, nodes.type =1, scenario =3) tree_beta$sp.list$genus <-gsub("Entada", "Gwilymia", tree_beta$sp.list$genus) tree_beta$sp.list$species <-gsub("Entada", "Gwilymia", tree_beta$sp.list$species) tree_beta$phylo$tip.lab <-gsub("Entada", "Gwilymia", tree_beta$phylo$tip.lab) tree_beta$sp.list$genus <-gsub("Enterolobium", "Robrichia", tree_beta$sp.list$genus) tree_beta$sp.list$species <-gsub("Enterolobium", "Robrichia", tree_beta$sp.list$species) tree_beta$phylo$tip.lab <-gsub("Enterolobium", "Robrichia", tree_beta$phylo$tip.lab) tree_beta$sp.list$species[which(tree_beta$sp.list$species %in% splist_cor$species[which(Species_list$genus =="Marlimorimia")])] <- Species_list$species[which(Species_list$genus =="Marlimorimia")] tree_beta$sp.list$genus[which(tree_beta$sp.list$species %in% splist_cor$species[which(Species_list$genus =="Marlimorimia")])] <-"Marlimorimia" tree_beta$phylo$tip.lab[which(tree_beta$phylo$tip.lab %in%gsub(" ", "_", splist_cor$species[which(Species_list$genus =="Marlimorimia")]))] <- Species_list$species[which(Species_list$genus =="Marlimorimia")] tree_beta$phylo$tip.label <- tree_beta$phylo$tip.labsaveRDS(tree_beta, here_rel("data", "inventory_data", "tree_beta.rds"))}tree_beta$phylo$tip.lab <-gsub("_", " ", tree_beta$phylo$tip.lab)if (file.exists(here_rel("results", "models", "H1", "phylodata_H1.rds")) &file.exists(here_rel("results", "models", "H1", "crlgmulti_H1.rds"))) { phylodata <-readRDS(here_rel("results", "models", "H1", "phylodata_H1.rds")) crlgmulti <-readRDS(here_rel("results", "models", "H1", "crlgmulti_H1.rds"))} else { phylodata <- phylobase::phylo4d(tree_beta$phylo, data.frame(species =gsub("_", " ", tree_beta$phylo$tip.lab)) |>left_join(Summary_beta)) phylodata@data$species <-NULLsaveRDS(phylodata, here_rel("results", "models", "H1", "phylodata_H1.rds")) crlgmulti <- parallel::mclapply(colnames(phylodata@data), \(i){ phylosignal::phyloCorrelogram(phylodata, trait = i, ci.bs =1000) },mc.cores =10 )setNames(crlgmulti, colnames(phylodata@data)) -> crlgmultisaveRDS(crlgmulti, here_rel("results", "models", "H1", "crlgmulti_H1.rds"))}
We constructed a phylogeny using the GBOTB megatree as a backbone and attached missing taxa using U.PhyloMaker (scenario 3). Because a small number of genera have alternative circumscriptions between the French Guiana checklist and the megatree, we applied explicit name mappings (e.g., Marlimorimia ↔︎ Inga) to ensure consistent tip matching. We then assembled a phylo4d object that links each species (tip) to its set of environmental preference coefficients.
Table 1: Phylogenetic signal of species environmental preferences.
parameter
Lambda
Clim1
0.1177 (p=0.001)
Clim2
0.264 (p=0.001)
GeomSub02
0.0603 (p=0.0152)
GeomSub03
0.0297 (p=0.2579)
GeomSub04
0.0821 (p=0.001)
GeomSub05
0.016 (p=0.3174)
GeomSub06
0.0201 (p=0.4328)
GeomSub07
0.17 (p=0.001)
GeomSub08
0.0519 (p=0.0216)
GeomSub09
0.1122 (p=0.001)
GeomSub10
0.0286 (p=0.0782)
GeomSub11
1e-04 (p=1)
GeomSub12
0.4133 (p=0.001)
GeomSub13
0.0886 (p=0.19)
logSWI
0.1555 (p=0.001)
We quantify phylogenetic signal in each coefficient using Pagel’s λ. This statistic measures the extent to which trait variation across species covaries with shared evolutionary history under a Brownian-motion reference model. Values near 0 indicate weak phylogenetic structure (preferences vary independently of phylogeny), whereas higher values indicate that related species tend to share similar preferences.
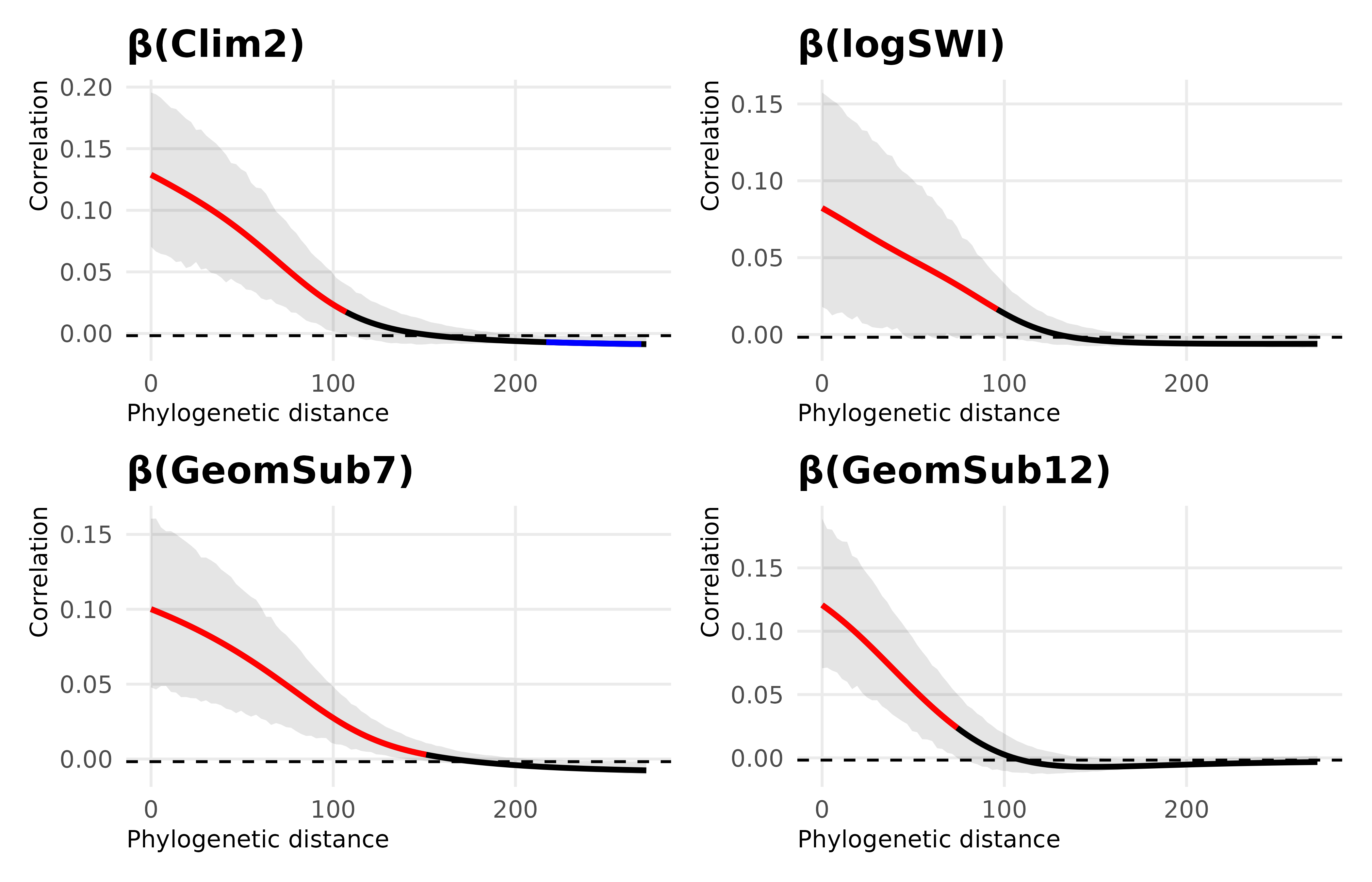
Figure 1: Phylogenetic correlograms for species‐level environmental coefficients with significant phylogenetic signal detected (Clim2, SWI, complex hilly landscape and high hills landscape)
Global λ values summarise phylogenetic signal across the entire tree, but they do not indicate at which phylogenetic depths similarity is strongest. To address this, we compute phylogenetic correlograms, which quantify trait autocorrelation as a function of patristic (branch-length) distance. This allows us to distinguish short-range conservatism among close relatives from deeper, clade-level patterns and potential long-distance opposition.
Regional climate
A moderate significant phylogenetic signal was detected for \(Clim_2\) (Pagel’s λ = 0.26), with a positive phylogenetic autocorrelation at short patristic distances, meaning that phylogenetically close species are more likely to prefer the same climatic condition defined by \(Clim_2\), and a negative association at long distancesFigure 1.
Geomorphological landscapes
Phylogenetic signals were significant for “\(GeomSub_{[12]}\) high hills” (λ = 0.41, p < 0.001) and “\(GeomSub_{[7]}\) complex hilly area” (λ = 0.17, p = 0.001). Both exhibited positive autocorrelation at short patristic distancesFigure 1. The pattern was negative at long patristic distance for “\(GeomSub_{[7]}\) complex hilly areas” and non-significant for “\(GeomSub_{[12]}\) high hills” Figure 1.
Local topography
A significant phylogenetic signal was detected for \(SWI\) (λ = 0.16, p < 0.001) with a positive phylogenetic autocorrelation at short patristic distances and non-significant at long patristic distances.
While correlograms describe average autocorrelation patterns, they do not identify where in the phylogeny the signal is concentrated. We therefore compute Local Indicators of Phylogenetic Association (LIPA), which provide a tip- and clade-specific measure of whether a species’ coefficient value is unusually similar to (positive LIPA) or unusually different from (negative LIPA) those of its phylogenetic neighbours, relative to a permutation-based null model.
LIPA outputs are interpreted as local departures from the global phylogenetic expectation. Positive values highlight phylogenetic clustering of preferences (local conservatism), whereas negative values highlight local overdispersion (close relatives differing more than expected). We visualise these patterns on the phylogeny for the coefficients that exhibited significant global signal.
Regional climate
We focus on the regional climate Clim2, which showed significant phylogenetic structure. The figure below maps LIPA values onto the phylogeny to identify which clades contribute most strongly to the detected signal.
LIPA associated to regional climate highlighted positive signals for Sapotaceae and for the genera Eschweilera, Lecythis, Tovomita, Rinorea, Goupia, Eperua, and ProtiumFigure 2.
Geomophological landscapes
We then repeat the same LIPA-based localisation for geomorphological subtypes that exhibit significant phylogenetic signal. Because geomorphological subtypes represent mesoscale hydro-edaphic contexts, clade-level clustering can be interpreted as lineage-specific habitat affinities associated with soil development, drainage constraints, and related functional strategies.
LIPA associated to “\(GeomSub_{[12]}\) high hills” geomorphological landscapes revealed positive signals located in Eschweilera, Lecythis and LicaniaFigure 4.
Local topography
Finally, we assess whether local topographic preferences (SWI coefficient) are phylogenetically clustered or dispersed. Because SWI captures fine-scale variation in drainage and waterlogging expose, local dispersion within genera would be consistent with repeated ecological divergence among close relatives along local topographic gradients.
LIPA associated to local topography identified positive signals in Sapotaceae and in the genera Eschweilera, Inga, Vismia, and Carapa, and negative signals in Eschweilera, Pouteria, Symphonia, and ZygiaFigure 5.
Within-genus specialisation along SWI gradient
Global phylogenetic signal can coexist with recent divergence within genera. To characterise potential evolutionary differentiation, we centre each species’ SWI coefficient relative to its genus mean. This highlights congeneric species that consistently deviate from their genus-average position along the SWI gradient, which is interpreted here as within-genus specialisation.
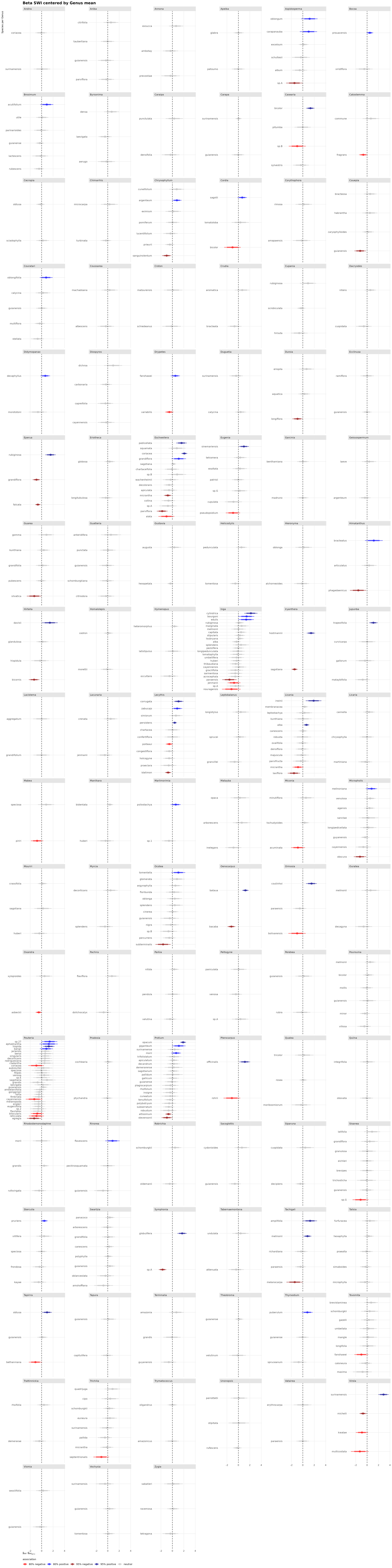
Figure 6: Phylogenetic tree of the studied specie selection among identified tree and palm species in French Guiana
The genus-centred coefficients summarise whether species are systematically shifted toward wetter or drier microtopographic positions relative to their closest congeners. We interpret consistent positive or negative deviations (with credible intervals excluding zero) as evidence for within-genus differentiation in microtopographic affinity, which may contribute to local coexistence by reducing niche overlap among close relatives.
By testing species preference differences within genera, within-genus comparison, we found that 25 species out of 105 multispecies genera, showed opposite SWI affinities to at least one congener.
Source Code
---title: "Phylogenetic analysis"format: html: code-fold: true---```{r libraries-data, warning=FALSE, message=FALSE, include=FALSE}library(tidyverse)library(U.PhyloMaker)library(gt)library(gtExtras)library(patchwork)here_rel <- function(...) { fs::path_rel(here::here(...))}source(here_rel("R", "funs_data.R"))source(here_rel("R", "funs_graphics.R"))source(here_rel("R", "funs_plot_tables.R"))theme_set(theme_public())```# OverviewThis section evaluates whether species’ environmental preferences inferred from the JSDM are phylogenetically structured. We first quantify global phylogenetic signal in each species-level coefficient using Pagel’s λ, then characterise how this signal varies across phylogenetic distances using phylogenetic correlograms. Finally, we use Local Indicators of Phylogenetic Association (LIPA) to identify clades in which closely related species show unusually similar (or unusually dissimilar) preferences.To relate model outputs to evolutionary history, we combine two data sources: (i) posterior summaries of species-specific environmental coefficients (regional climate, geomorphological subtypes, and SWI), and (ii) a backbone seed-plant phylogeny. The workflow below harmonises taxonomy between the French Guiana checklist and the megatree, constructs a phylogeny for the analysed species, and matches each tip to the corresponding coefficient values.```{r PhyloSignal}Species_list_path <- here_rel("data", "inventory_data", "Species_list.csv")Summary_beta <- vroom::vroom(here_rel("results", "models", "H1", "beta_fitH1.tsv.gz")) |> filter(Scale %in% c("Regional", "Landscape", "Local")) |> mutate( # significance at 80% CI sig80 = (l > 0 & h > 0) | (l < 0 & h < 0), sign = case_when( sig80 & m > 0 ~ "positive", sig80 & m < 0 ~ "negative", TRUE ~ "ns" ) ) |> mutate(species = paste(genus, species)) |> select(species, m, parameter) |> spread(parameter, m)Species_list <- read_csv2(Species_list_path) |> rename(SpEpithet = specie, species = taxonomic_name) |> select(species, genus, family, selected) |> filter(species %in% Summary_beta$species, !duplicated(species))if (file.exists(here_rel("data", "inventory_data", "tree_beta.rds"))) { tree_beta <- readRDS(here_rel("data", "inventory_data", "tree_beta.rds"))} else { megatree <- read.tree("https://github.com/megatrees/plant_20221117/raw/refs/heads/main/plant_megatree.tre") gen.list <- read.csv("https://github.com/megatrees/plant_20221117/raw/refs/heads/main/plant_genus_list.csv", sep = ",") # Correction for modified genus between megatree / species list (Molino et al. 2022). splist_cor <- Species_list splist_cor$genus <- gsub("Marlimorimia", "Inga", splist_cor$genus) splist_cor$species <- gsub("Marlimorimia", "Inga", splist_cor$species) splist_cor$genus <- gsub("Robrichia", "Enterolobium", splist_cor$genus) splist_cor$species <- gsub("Robrichia", "Enterolobium", splist_cor$species) splist_cor$genus <- gsub("Gwilymia", "Entada", splist_cor$genus) splist_cor$species <- gsub("Gwilymia", "Entada", splist_cor$species) tree_beta <- phylo.maker(splist_cor, megatree, gen.list, nodes.type = 1, scenario = 3) tree_beta$sp.list$genus <- gsub("Entada", "Gwilymia", tree_beta$sp.list$genus) tree_beta$sp.list$species <- gsub("Entada", "Gwilymia", tree_beta$sp.list$species) tree_beta$phylo$tip.lab <- gsub("Entada", "Gwilymia", tree_beta$phylo$tip.lab) tree_beta$sp.list$genus <- gsub("Enterolobium", "Robrichia", tree_beta$sp.list$genus) tree_beta$sp.list$species <- gsub("Enterolobium", "Robrichia", tree_beta$sp.list$species) tree_beta$phylo$tip.lab <- gsub("Enterolobium", "Robrichia", tree_beta$phylo$tip.lab) tree_beta$sp.list$species[which(tree_beta$sp.list$species %in% splist_cor$species[which(Species_list$genus == "Marlimorimia")])] <- Species_list$species[which(Species_list$genus == "Marlimorimia")] tree_beta$sp.list$genus[which(tree_beta$sp.list$species %in% splist_cor$species[which(Species_list$genus == "Marlimorimia")])] <- "Marlimorimia" tree_beta$phylo$tip.lab[which(tree_beta$phylo$tip.lab %in% gsub(" ", "_", splist_cor$species[which(Species_list$genus == "Marlimorimia")]))] <- Species_list$species[which(Species_list$genus == "Marlimorimia")] tree_beta$phylo$tip.label <- tree_beta$phylo$tip.lab saveRDS(tree_beta, here_rel("data", "inventory_data", "tree_beta.rds"))}tree_beta$phylo$tip.lab <- gsub("_", " ", tree_beta$phylo$tip.lab)if (file.exists(here_rel("results", "models", "H1", "phylodata_H1.rds")) & file.exists(here_rel("results", "models", "H1", "crlgmulti_H1.rds"))) { phylodata <- readRDS(here_rel("results", "models", "H1", "phylodata_H1.rds")) crlgmulti <- readRDS(here_rel("results", "models", "H1", "crlgmulti_H1.rds"))} else { phylodata <- phylobase::phylo4d(tree_beta$phylo, data.frame(species = gsub("_", " ", tree_beta$phylo$tip.lab)) |> left_join(Summary_beta)) phylodata@data$species <- NULL saveRDS(phylodata, here_rel("results", "models", "H1", "phylodata_H1.rds")) crlgmulti <- parallel::mclapply(colnames(phylodata@data), \(i){ phylosignal::phyloCorrelogram(phylodata, trait = i, ci.bs = 1000) }, mc.cores = 10 ) setNames(crlgmulti, colnames(phylodata@data)) -> crlgmulti saveRDS(crlgmulti, here_rel("results", "models", "H1", "crlgmulti_H1.rds"))}```We constructed a phylogeny using the GBOTB megatree as a backbone and attached missing taxa using U.PhyloMaker (scenario 3). Because a small number of genera have alternative circumscriptions between the French Guiana checklist and the megatree, we applied explicit name mappings (e.g., Marlimorimia ↔ Inga) to ensure consistent tip matching. We then assembled a phylo4d object that links each species (tip) to its set of environmental preference coefficients.```{r}#| label: tbl-phylosignal#| tbl-cap: "Phylogenetic signal of species environmental preferences."phylosignal::phyloSignal(p4d = phylodata, method ="Lambda") |>lapply(as.data.frame) |>lapply(rownames_to_column, "parameter") |>bind_rows(.id ="type") |> reshape2::melt(c("type", "parameter")) |> reshape2::dcast(parameter + variable ~ type) |>mutate(parameter =if_else(substr(parameter,1,2) =="Ge", paste0("GeomSub", str_pad(gsub("GeomSub", "", parameter),width =2,side ="left", pad ="0" )), parameter)) |>mutate(value =paste0(round(stat, 4), " (p=", round(pvalue, 4), ")")) |> reshape2::dcast(parameter ~ variable) |> knitr::kable()``` We quantify phylogenetic signal in each coefficient using Pagel’s λ. This statistic measures the extent to which trait variation across species covaries with shared evolutionary history under a Brownian-motion reference model. Values near 0 indicate weak phylogenetic structure (preferences vary independently of phylogeny), whereas higher values indicate that related species tend to share similar preferences.```{r warning=FALSE, message=FALSE}#| label: fig-crlg#| fig-cap: "Phylogenetic correlograms for species‐level environmental coefficients with significant phylogenetic signal detected (Clim2, SWI, complex hilly landscape and high hills landscape)"(plot_crlg(crlgmulti, "Clim2") | plot_crlg(crlgmulti, "logSWI")) / (plot_crlg(crlgmulti, "GeomSub7") | plot_crlg(crlgmulti, "GeomSub12")) + plot_layout(guides = "collect") + theme_public()```Global λ values summarise phylogenetic signal across the entire tree, but they do not indicate at which phylogenetic depths similarity is strongest. To address this, we compute phylogenetic correlograms, which quantify trait autocorrelation as a function of patristic (branch-length) distance. This allows us to distinguish short-range conservatism among close relatives from deeper, clade-level patterns and potential long-distance opposition.## Regional climateA moderate significant phylogenetic signal was detected for $Clim_2$ (Pagel’s λ = 0.26), with a **positive phylogenetic autocorrelation** at **short patristic distances**, meaning that phylogenetically close species are more likely to prefer the same climatic condition defined by $Clim_2$, and a **negative** association at **long distances** @fig-crlg. ## Geomorphological landscapesPhylogenetic signals were significant for “$GeomSub_{[12]}$ high hills” (λ = 0.41, p < 0.001) and “$GeomSub_{[7]}$ complex hilly area” (λ = 0.17, p = 0.001). Both exhibited **positive autocorrelation** at **short patristic distances** @fig-crlg. The pattern was **negative** at **long patristic distance** for “$GeomSub_{[7]}$ complex hilly areas” and **non-significant** for “$GeomSub_{[12]}$ high hills” @fig-crlg. ## Local topography A significant phylogenetic signal was detected for $SWI$ (λ = 0.16, p < 0.001) with a **positive** phylogenetic autocorrelation at **short patristic distances** and **non-significant** at **long patristic distances**.# Local phylogenetic association indicators (LIPA)```{r warning=FALSE, message=FALSE}if (file.exists(here_rel("results", "models", "H1", "lipa_beta_H1.rds"))) { lipa_beta <- readRDS(here_rel("results", "models", "H1", "lipa_beta_H1.rds"))} else { lipa_beta <- phylosignal::lipaMoran(phylodata, alternative = "two-sided", reps = 9999, prox.phylo = "patristic") saveRDS(lipa_beta, here_rel("results", "models", "H1", "lipa_beta_H1.rds"))}```While correlograms describe average autocorrelation patterns, they do not identify where in the phylogeny the signal is concentrated. We therefore compute Local Indicators of Phylogenetic Association (LIPA), which provide a tip- and clade-specific measure of whether a species’ coefficient value is unusually similar to (positive LIPA) or unusually different from (negative LIPA) those of its phylogenetic neighbours, relative to a permutation-based null model.LIPA outputs are interpreted as local departures from the global phylogenetic expectation. Positive values highlight phylogenetic clustering of preferences (local conservatism), whereas negative values highlight local overdispersion (close relatives differing more than expected). We visualise these patterns on the phylogeny for the coefficients that exhibited significant global signal.## Regional climateWe focus on the regional climate Clim2, which showed significant phylogenetic structure. The figure below maps LIPA values onto the phylogeny to identify which clades contribute most strongly to the detected signal.```{r warning=FALSE, message=FALSE}#| label: fig-LIPA-Clim2if (!file.exists(here_rel("notebook", "figs", "LIPA_H1_Clim2.png"))) { g <- plot_LIPA(lipa_beta, phylodata, "Clim2") ggsave(plot = g, filename = here_rel("notebook", "figs", "LIPA_H1_Clim2.png"), bg = "white", width = 20, height = 20, dpi = 1000)}knitr::include_graphics(here_rel("notebook", "figs", "LIPA_H1_Clim2.png"))```LIPA associated to regional climate highlighted **positive signals** for *Sapotaceae* and for the genera *Eschweilera*, *Lecythis*, *Tovomita*, *Rinorea*, *Goupia*, *Eperua*, and *Protium* @fig-LIPA-Clim2.## Geomophological landscapesWe then repeat the same LIPA-based localisation for geomorphological subtypes that exhibit significant phylogenetic signal. Because geomorphological subtypes represent mesoscale hydro-edaphic contexts, clade-level clustering can be interpreted as lineage-specific habitat affinities associated with soil development, drainage constraints, and related functional strategies.### Complex hilly```{r warning=FALSE, message=FALSE}#| label: fig-LIPA-GeomSub07if (!file.exists(here_rel("notebook", "figs", "LIPA_H1_GeomSub7.png"))) { g <- plot_LIPA(lipa_beta, phylodata, "GeomSub7") ggsave( plot = g, filename = here_rel("notebook", "figs", "LIPA_H1_GeomSub7.png"), bg = "white", width = 20, height = 20, dpi = 1000 )}knitr::include_graphics(here_rel("notebook", "figs", "LIPA_H1_GeomSub7.png"))```IPA associated to “$GeomSub_{[7]}$ Complex hilly” geomorphological landscapes revealed **positive signals** located in *Eschweilera* and *Lecythis* @fig-LIPA-GeomSub07.### high hills```{r warning=FALSE, message=FALSE}#| label: fig-LIPA-GeomSub12if (!file.exists(here_rel("notebook", "figs", "LIPA_H1_GeomSub12.png"))) { g <- plot_LIPA(lipa_beta, phylodata, "GeomSub12") ggsave( plot = g, filename = here_rel("notebook", "figs", "LIPA_H1_GeomSub12.png"), bg = "white", width = 20, height = 20, dpi = 1000 )}knitr::include_graphics(here_rel("notebook", "figs", "LIPA_H1_GeomSub12.png"))```LIPA associated to “$GeomSub_{[12]}$ high hills” geomorphological landscapes revealed **positive signals** located in *Eschweilera*, *Lecythis* and *Licania* @fig-LIPA-GeomSub12.## Local topographyFinally, we assess whether local topographic preferences (SWI coefficient) are phylogenetically clustered or dispersed. Because SWI captures fine-scale variation in drainage and waterlogging expose, local dispersion within genera would be consistent with repeated ecological divergence among close relatives along local topographic gradients.```{r warning=FALSE, message=FALSE}#| label: fig-LIPA-SWIif (!file.exists(here_rel("notebook", "figs", "LIPA_H1_SWI.png"))) { g <- plot_LIPA(lipa_beta, phylodata, "logSWI") ggsave( plot = g, filename = here_rel("notebook", "figs", "LIPA_H1_SWI.png"), bg = "white", width = 20, height = 20, dpi = 1000 )}knitr::include_graphics(here_rel("notebook", "figs", "LIPA_H1_SWI.png"))```LIPA associated to local topography identified **positive signals** in *Sapotaceae* and in the genera *Eschweilera*, *Inga*, *Vismia*, and *Carapa*, and **negative signals** in *Eschweilera*, *Pouteria*, *Symphonia*, and *Zygia* @fig-LIPA-SWI. # Within-genus specialisation along SWI gradientGlobal phylogenetic signal can coexist with recent divergence within genera. To characterise potential evolutionary differentiation, we centre each species’ SWI coefficient relative to its genus mean. This highlights congeneric species that consistently deviate from their genus-average position along the SWI gradient, which is interpreted here as within-genus specialisation.```{r warning=FALSE, message=FALSE}#| label: fig-intra-genus#| fig-cap: "Phylogenetic tree of the studied specie selection among identified tree and palm species in French Guiana"if(!file.exists(here_rel("notebook", "figs", "div_intragenus_H1.png"))){ g <- vroom::vroom(here_rel("results", "models", "H1", "beta_fitH1.tsv.gz")) |> filter(parameter == "logSWI") |> mutate(specieslong = paste(genus,species))|> group_by(genus) |> reframe(specieslong, species, TWI_mean_genus = (m - mean(m)), l = (l - mean(m)), h = (h - mean(m)), ll = (ll - mean(m)), hh = (hh - mean(m))) |> filter( TWI_mean_genus != 0) |> mutate(association = "neutral") |> mutate(association = ifelse(l > 0 & h > 0, "80% positive", association)) |> mutate(association = ifelse(l < 0 & h < 0, "80% negative", association)) |> mutate(association = ifelse(ll > 0 & hh > 0, "95% positive", association)) |> mutate(association = ifelse(ll < 0 & hh < 0, "95% negative", association)) |> ggplot(aes(x = reorder(species, TWI_mean_genus, mean), xend = reorder(species, TWI_mean_genus, mean), color = association, fill = association)) + geom_point(aes(y = TWI_mean_genus), shape = 21, size = 3, alpha = 0.5) + coord_flip() + ylab(expression(beta[SWI] - beta[SWI[genus]] )) + geom_segment(aes(y = ll, yend = hh), linewidth = 1, show.legend = F, alpha = 0.5 ) + geom_segment(aes(y = l, yend = h), linewidth = 2, alpha = 0.5) + scale_color_manual(values = c("95% positive" = "darkblue", "80% positive" = "blue", "neutral" = "grey", "80% negative" = "red", "95% negative" = "darkred")) + scale_fill_manual(values = c("95% positive" = "darkblue", "80% positive" = "blue", "neutral" = "grey", "80% negative" = "red", "95% negative" = "darkred")) + geom_hline(yintercept = 0, col = "black", linetype = "dashed") + theme( axis.title.y = element_blank(), axis.text.y = element_text(face = "italic"), strip.text = element_text(face = "italic"), legend.position = "bottom" ) + facet_wrap(~genus, scales = "free_y", ncol = 6) + xlab("Species per Genus") + ggtitle("Beta SWI centered by Genus mean") + theme_public() ggsave(plot = g, filename = here_rel("notebook", "figs","div_intragenus_H1.png"), bg = "white", width =20,height = 80, dpi = 300,limitsize = FALSE)} knitr::include_graphics(here_rel("notebook", "figs","div_intragenus_H1.png"))```The genus-centred coefficients summarise whether species are systematically shifted toward wetter or drier microtopographic positions relative to their closest congeners. We interpret consistent positive or negative deviations (with credible intervals excluding zero) as evidence for within-genus differentiation in microtopographic affinity, which may contribute to local coexistence by reducing niche overlap among close relatives.By testing species preference differences within genera, within-genus comparison, we found that 25 species out of 105 multispecies genera, showed opposite SWI affinities to at least one congener.